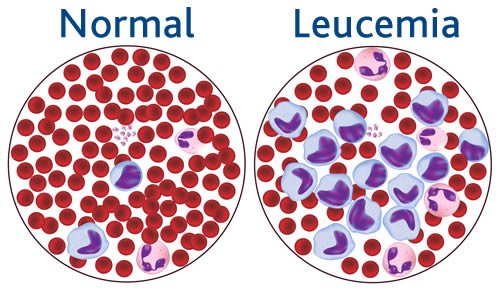
La leucemia linfoblastica acuta (LLA) è una malattia oncoematologica che coinvolge il sangue e il midollo osseo, il tessuto che si trova all’interno delle ossa, da cui hanno origine le cellule del sangue periferico.
È una patologia rara, a rapida progressione. La malattia coinvolge le cellule ematologiche immature bloccandone la differenziazione.
Si verifica quando all’interno di una cellula del midollo osseo avviene una mutazione o un errore nella duplicazione del DNA che ne altera i normali processi di proliferazione e differenziazione. Nei pazienti con LLA, avviene un accumulo incontrollato di globuli bianchi immaturi e maligni (detti blasti) che toglie spazio alle cellule sane del midollo osseo, 2 compromettendo le normali funzioni ematopoietiche.
Incidenza
In Europa, vengono diagnosticati complessivamente ogni anno 7 mila casi di LLA. Esistono vari sottotipi di LLA, con un’incidenza specifica molto bassa. Per esempio, si stima che in Francia, Germania, Italia, Spagna e Regno Unito siano circa 600 gli adulti con LLA da precursori delle cellule B recidivante o refrattaria negativa per il cromosoma Philadelphia. I pazienti adulti con diagnosi di LLA sono spesso giovani adulti, con un’età media alla diagnosi di 44-55 anni.
Sintomi
In generale, la LLA si manifesta clinicamente con:
- Sanguinamento delle gengive
- Febbre
- Infezioni frequenti
- Sanguinamento dal naso, abbondante e frequente
- Riscontro di linfonodi ingrossati, sul collo, sotto le ascelle, a livello addominale o all’inguine
- Pallore
- Respiro corto
- Debolezza, affaticamento.
Fattori di rischio
Non si conoscono con certezza le cause che provocano le mutazioni del DNA responsabili della produzione di cellule linfoidi anomale e quindi della LLA, ma si sa che la malattia non è ereditaria.
I fattori che possono aumentare il rischio di LLA sono:
- Precedenti terapie contro il cancro
- Esposizione a radiazioni
- Sindromi genetiche
- Familiarità per LLA.
DIAGNOSI
I test e le procedure effettuabili per la diagnosi della LLA includono:
- Analisi del sangue: le analisi del sangue potrebbero rivelare anomalie nell’emocromo come, ad esempio, un elevato numero di globuli bianchi, bassi livelli di globuli rossi e di piastrine. Le analisi del sangue mostrano anche la presenza di blasti (cellule immature) che si trovano solitamente nel midollo osseo, ma che non circolano nel sangue.
- Analisi del midollo osseo: attraverso l’utilizzo di un ago, viene prelevato un campione del midollo osseo dall’osso del bacino, per ricercare le cellule leucemiche. I medici, in laboratorio, classificano le cellule ematopoietiche in base alla loro dimensione, alla forma e altre caratteristiche. Cercano anche di individuare alcuni cambiamenti tipici delle cellule patologiche e di determinare se le cellule leucemiche derivano da linfociti B o linfociti T.
- Test di imaging: i test di imaging, come le radiografie del torace, la tomografia computerizzata (TC) o l’ecografia possono aiutare a stabilire se la malattia coinvolge il cervello, le ossa, i linfonodi o altre parti del corpo.
- Analisi del liquido cefalorachidiano (liquor): attraverso una puntura lombare, si raccoglie un campione di liquor. Il campione viene analizzato alla ricerca dell’eventuale presenza di cellule leucemiche.
Ci sono vari sottotipi di LLA, la cui corretta identificazione è importante per una corretta diagnosi e fondamentale per la successiva scelta terapeutica.
Attraverso l’analisi dell’immunofenotipo delle cellule leucemiche è possibile distinguere i due gruppi principali di LLA: la LLA di origine B (la più comune) e la LLA di origine T.
È possibile inoltre classificare la LLA in base allo stadio maturativo a cui sono assimilabili i blasti leucemici e alla presenza o meno del cromosoma Philadelphia.
TRATTAMENTI
Solitamente, il trattamento della LLA si compone di più fasi di chemioterapia:
- Fase d’induzione: lo scopo di questa prima fase della cura è quello di eliminare la maggior parte delle cellule leucemiche nel sangue e nel midollo osseo, e favorire il ripristino di cellule normali.
- Fase di consolidamento: la fase di consolidamento è nota anche come “terapia post-remissione”. Questa fase del trattamento mira a distruggere le cellule leucemiche residue e non individuabili.
- Fase di mantenimento: ha lo scopo di prevenire la ricomparsa di nuove cellule leucemiche e quindi la recidiva di malattia. Le dosi di chemioterapia somministrate in questa fase sono, solitamente, minori.
- Profilassi del Sistema Nervoso Centrale: i pazienti con LLA vengono sottoposti a trattamenti in grado di eliminare l’eventuale presenza di cellule leucemiche nel sistema nervoso centrale, durante tutte le fasi della terapia. Per questo tipo di trattamento, vengono iniettati farmaci chemioterapici direttamente nel canale spinale con lo scopo di uccidere le cellule leucemiche che non vengono raggiunte in questa sede dai normali farmaci chemioterapici assunti per via orale, iniettati sottopelle (per via sottocutanea) o via endovenosa.
Circa l’11% dei pazienti non risponde alla cura, e soffre della cosiddetta leucemia refrattaria. Circa il 60% dei pazienti affetti da LLA va incontro invece a ricaduta dopo aver inizialmente risposto al trattamento. Per i pazienti adulti con LLA, la probabilità di sopravvivenza a cinque anni, dopo la prima ricaduta, è del 7%. La mortalità è quindi altissima.
In relazione alla severità del quadro clinico e all’eventuale progressione della malattia, le cure della LLA possono durare due o tre anni, e prevedono:
- Chemioterapia: ovvero farmaci che uccidono le cellule leucemiche e, solitamente, ciò avviene durante la fase di induzione sia nei bambini che negli adulti; può essere anche utilizzata durante la fase di consolidamento e mantenimento.
- Terapia mirata: farmaci che colpiscono specificamente alcune anomalie delle cellule leucemiche, che sono alla base della loro sopravvivenza e proliferazione illimitata.
- Radioterapia: utilizzo di radiazioni, che uccidono le cellule tumorali.
- Trapianto di cellule staminali: può essere utilizzato durante la terapia di consolidamento nei pazienti a rischio di ricaduta per ristabilire la presenza di cellule staminali sane, che sostituiscano quelle tumorali presenti nel midollo osseo e instaurare una risposta immunologica contro la leucemia.
- Sperimentazioni cliniche: testano l’efficacia delle nuove cure per la leucemia o quella delle terapie esistenti. Le sperimentazioni cliniche danno una possibilità ai pazienti di essere trattati con farmaci innovativi, per i quali sono ancora in corso di valutazione i rischi e i benefici. I pazienti discutono i potenziali rischi e benefici con il loro medico, prima di firmare un consenso informato per essere inseriti all’interno della sperimentazione clinica.
Lo scopo della cura è la remissione completa (RC). La remissione avviene quando non ci sono più evidenze di LLA e sia l’emocromo che le cellule del midollo osseo del paziente ritornano nella norma. Nella LLA, la remissione è spesso definita da un numero di blasti leucemici nel midollo osseo inferiore al 5%.
Test MRD
Con il raggiungimento della RC, potrebbe essere presente, nell’organismo, un piccolo numero di cellule leucemiche non visibili. Si definisce residuo minimo di malattia (MRD): uno stato in cui la leucemia non è visibile al microscopio, ma solo attraverso tecniche più sensibili in grado di individuare le cellule maligne residue.
Il test MRD può essere utilizzato per valutare la prognosi dei pazienti con LLA e per guidare le decisioni sull’iter di trattamento. La valutazione della MRD ha acquisito un valore importante nei protocolli di cura europei per i pazienti con LLA, sulla base del suo alto valore prognostico.
“Lo strumento terapeutico di riferimento rimane la chemioterapia, che si basa su farmaci che sono stati sviluppati a partire dagli anni ’60 – precisa Alessandro Rambaldi, Direttore Unità Strutturale Complessa di Ematologia, Azienda ASST Papa Giovanni XXIII Bergamo –. Nel corso degli anni abbiamo imparato ad usare sempre meglio questi farmaci definendo le combinazioni tra i chemioterapici, la loro sequenza, e soprattutto la dose di utilizzo e questo ha portato già grandi risultati: i bambini trattati negli anni ’70 con gli stessi farmaci che utilizziamo oggi guarivano nel 20% dei casi, oggi nel 90% dei casi. Nel paziente adulto il progresso è stato meno significativo, ma comunque molto rilevante. Il secondo presidio terapeutico disponibile è il trapianto di midollo osseo allogenico che è una modalità estremamente efficace perché si basa sulla somministrazione di una dose massimale di chemioterapia eventualmente associata anche alla radioterapia. Questa combinazione da un lato è in grado di ottenere una significativa eradicazione della malattia, ma al tempo stesso provoca un danno irreversibile anche alle cellule sane che non sono più in grado di produrre sangue. Per tale ragione questa funzione viene assunta da cellule staminali emopoietiche sane ottenute da un donatore compatibile. Purtroppo, accanto all’effetto terapeutico del trapianto, il sistema immunitario del donatore può talvolta aggredire non soltanto le cellule leucemiche, ma anche quelle sane del paziente (quali quelle della pelle, dell’intestino, del fegato e determinare una malattia che si chiama ‘malattia del trapianto contro l’ospite’, Graft Versus Host Disease, GvHD). Questa complicanza interessa circa il 25% dei pazienti e si presenta con delle manifestazioni acute o croniche che possono compromettere la qualità di vita del paziente e qualche volta la vita stessa. Per tutte queste ragioni, il trapianto di midollo osseo allogenico viene offerto solo ai pazienti per i quali si ha la certezza o almeno un’alta probabilità che la chemioterapia da sola non possa portare a guarigione.
Grazie al trapianto e a tanta ricerca di laboratorio, nel corso degli ultimi 35 anni abbiamo capito che una terapia immunologica contro la leucemia era possibile ed estremamente efficace. Dobbiamo sfruttare il sistema immunitario per curare in modo definitivo questa malattia senza però causare molte delle tossicità prima descritte. Tra questi nuovi trattamenti va ricordato lo sviluppo di tecniche di terapia genica, grazie alle quali i linfociti T vengono geneticamente modificati per riconoscere bersagli espressi sulla superficie delle cellule leucemiche o lo sviluppo di nuovi anticorpi, che ottengono lo stesso risultato attivando i linfociti stessi del paziente.
Da questi nuovi trattamenti innovativi più efficaci e meno tossici rispetto alla chemioterapia e al trapianto di midollo osseo ci aspettiamo nuovi decisivi progressi nella terapia di questa malattia”.
Sulle cure innovative risponde Robin Foà Direttore Ematologia, Policlinico Umberto I, Sapienza Università di Roma
Cosa sono gli anticorpi bispecifici? È ora disponibile anche in Italia blinatumomab, primo anticorpo bispecifico per il trattamento della leucemia acuta linfoblastica. Ci può spiegare in cosa consiste il suo innovativo meccanismo d’azione?
Il blinatumomab è un anticorpo monoclonale definito bispecifico perché il bersaglio è rappresentato da due antigeni espressi sulla superficie delle cellule, non uno come di solito avviene per gli anticorpi monoclonali. Uno è un antigene chiamato CD19 che è presente sulla superficie delle cellule di tutte le leucemie acute linfoblastiche della serie B, ma il meccanismo d’azione è mediato invece dall’altro antigene, il CD3, che è presente sulla superficie dei linfociti T del paziente. È una forma di immunoterapia perché di fatto vengono attivati/armati i linfociti T del paziente a riconoscere le cellule leucemiche CD19+. L’innovazione sta nel modello di azione; il blinatumomab è il primo anticorpo monoclonale bispecifico che è arrivato nella clinica ed è stato recentemente approvato prima da FDA e poi da EMA, e ora anche da AIFA.
La terapia con blinatumomab è quindi una forma di immunoterapia: viene attivato il sistema immunitario del paziente a riconoscere le cellule malate e quindi a cercare di eliminarle. È il primo anticorpo bispecifico approvato in oncologia e rappresenta una strategia terapeutica rivoluzionaria per una patologia molto grave. L’approvazione è per pazienti adulti che abbiano una leucemia acuta linfoblastica di tipo B, che sia Philadelphia (cioè che non porti questa alterazione citogenetica), e che siano recidivati o resistenti ad una o più linee di terapia. Quindi non si usa in prima linea ma nelle recidive di malattia.
Quali sono le evidenze scientifiche di efficacia di blinatumomab nel trattamento della leucemia linfoblastica acuta, in particolare rispetto alla chemioterapia, considerata finora il trattamento standard per questa patologia?
Sono stati condotti negli anni molti studi prima di arrivare allo studio TOWER randomizzato, che rappresenta l’ultimo anello di una lunga catena di studi clinici di fase I e II che sono stati completati in precedenza e che hanno dimostrato l’efficacia di questo anticorpo bispecifico in pazienti con leucemia linfoblastica acuta in recidiva o resistente alla terapia standard. I risultati incoraggianti hanno portato al disegno dello studio randomizzato per pazienti che hanno questo tipo di malattia, negativi per il cromosoma Philadelphia. I pazienti sono stati randomizzati alla cieca a ricevere lo standard of care, che è sempre una combinazione chemioterapica, oppure questo anticorpo monoclonale usato da solo. I risultati dello studio sono stati rilevanti perché hanno dimostrato che, rispetto alla terapia convenzionale, il blinatumomab ha permesso di ottenere percentuali di remissione completa di malattia significativamente più elevate e ha praticamente raddoppiato la sopravvivenza rispetto alla chemioterapia standard. Risultati quindi molto importanti, che non si erano mai osservati con un singolo farmaco. Questi risultati sono stati recentemente pubblicati sulla più prestigiosa rivista di medicina, il New England Journal of Medicine.
Per quanto tempo e con quali modalità viene somministrato blinatumomab ai pazienti?
Di solito si somministrano due cicli ma si può arrivare anche a cinque cicli. Le risposte maggiori si hanno dopo il primo e il secondo ciclo. Il trattamento consiste in una terapia infusionale continua, per 28 giorni, seguita da un periodo di intervallo di due settimane. Il paziente viene ricoverato all’inizio della terapia e successivamente la somministrazione del farmaco può proseguire a domicilio utilizzando una pompa da infusione.

